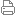
| |




姓名:D同学 背景: 国内普通高校 成绩:GPA88 托福115 GRE320 案例分析: D同学就读于国内普通高校的教育学专业,与 ...

AirServer Mac破解版,一款苹果Mac AirPlay屏幕镜像软件,专为Mac用户设计的Airplay终端实用工具,通过AirServer mac ...



R2D250-AF10-12 ebmpapst 散热离心风扇 类型 离心 型号 R2D250-AF10-12 电机功率 160(w) 电压 400(V) 电流 0.26 ...







目前玩剑盾1.3.2加两个dlc的没有问题,听说有钻石珍珠重置版,想下载,所以想问问吧友们11.0.0是不是玩不了钻石和珍珠 ...


苹果Mac Book air M1 2020款,8G运行内存+8GPU+512SSD内存,刚买小半年,几乎全新,在保, 自己办工用 ,电池循环次数 ...


随着情人节脚步临近,空气中弥漫着甜蜜的气息。京东MALL、京东电器城市旗舰店联手全国26座门店,发起温暖又隆重的“我 ...

11月份,规模以上工业主要能源产品生产同比均保持增长。其中,原煤增长4.6%,原油增长2.7%,天然气增长4.4%,发电量增 ...

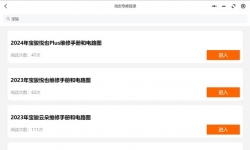
汽修帮手资料库提供各大厂家车型维修手册、电路图、新车特征、车身钣金维修数据、全车拆装、扭力、发动机大修、发动机 ...






🎬短剧《重生1980:开局卖茶叶蛋(80集)王雅清&胥宁轲》 🔗夸克网盘:https://pan.quark.cn/s/10cf9daec8e0 📖 简介 ...






亲爱的冒险者: 感谢大家公测以来对《终焉誓约》的关注与支持! 12月15日服务器已再次开放,并且今天上午我们也开放了 ...







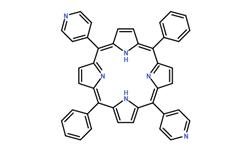
齐岳DPyP/cas71410-72-5/5,15-二(4-吡啶基)-10,20-二苯基卟啉) 英文名称:5,15-Di(4-Pyridyl)-10,20-Diphenylporphyri ...
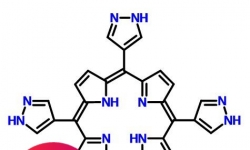
高纯cas1849676-26-1/5,10,15,20-四-1H-吡唑-4-基-21H,23H-卟啉 英文名称:5,10,15,20-tetra-pyrazol-4-yl porphyrin ...

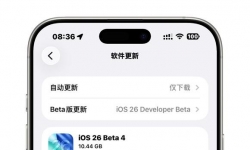
iPhone 16pro max beta3更新beta4升级包大小10.44g 1. 液态玻璃效果回归 •变化:Beta 3版本的“毛玻璃效果”被透明液 ...



基于bilibili@兔小美和你在一起的去除逝剑炸蛋版 Display:VMware SVGA ∥ 没有声音也没网,纯净 网页链接 ...








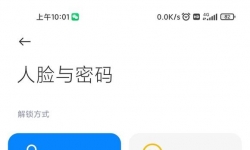
以前经常打开相机卡死黑屏一分多钟,恢复出厂设置好了几天,今天又出现了,卡屏以后指纹解锁就没用了,屏幕也没有指纹 ...





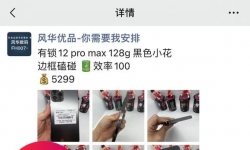
这里让自己更加了解 各种版本12 12pro 12pm行情 让自己少踩雷 回收行情也有的多避避坑 另外还有13系列 全部版本已经开 ...

新的数学题 开双卡 目前8q4 心悦懒的放半半 圣诞约一套。山巷大约会是多少玉,我有约数位染色的资本吗 妙笔又会是啥呜 ...



双卡5G用流量上网亮屏使用12个小时31分钟+双卡5G待机5个小时,续航可以, 红米NOTE11 Pro 8G+256G最新版本12.5.12.0测 ...









 微信扫一扫
微信扫一扫